ABSTRACT
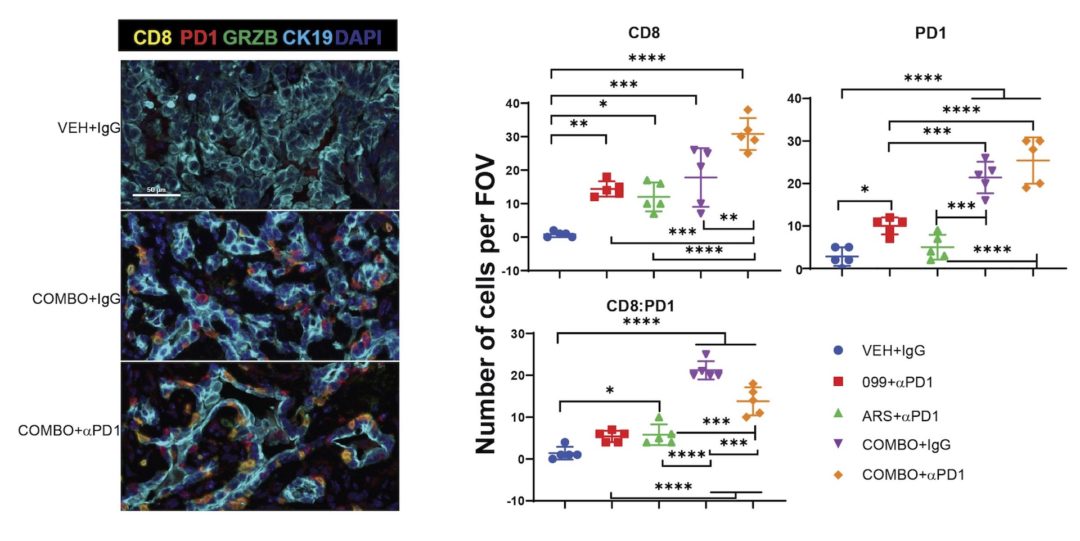
KRAS is the most frequently mutated oncogene in human cancer, and KRAS inhibition has been a longtime therapeutic goal. Recently, inhibitors (G12C-Is) that bind KRASG12C-GDP and react with Cys-12 were developed. Using new affinity reagents to monitor KRASG12C activation and inhibitor engagement, we found that, reflecting its action upstream of SOS1/2, SHP2 inhibitors (SHP2-Is) increased KRAS-GDP occupancy, enhancing G12C-I efficacy. SHP2-Is abrogated feedback signaling by multiple RTKs and blocked adaptive resistance to G12C-Is in vitro, in xenografts, and in syngeneic KRASG12C–mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) models. Biochemical analysis revealed enhanced suppression of ERK-, MYC-, anti-apoptotic-, and cell-cycle genes, and increased pro-apoptotic gene expression in tumors from combination-treated mice. SHP2-I/G12C-I also evoked favorable changes in the immune microenvironment, decreasing myeloid suppressor cells, increasing CD8+ T cells, and sensitizing tumors to PD-1 blockade. Experiments using cells expressing inhibitor-resistant SHP2 showed that SHP2 inhibition in PDAC cells is required for tumor regression and remodeling of the immune microenvironment, but also revealed direct inhibitory effects on angiogenesis resulting in decreased tumor vascularity. Our results demonstrate that SHP2-I/G12C-I combinations confer a substantial survival benefit in PDAC and NSCLC and identify additional combination strategies for enhancing the efficacy of G12C-Is.
[preprint posted on bioRxiv 2020 May 31]